| Acta Bioquímica Clínica Latinoamericana |
| Encefalopatía Espongiforme Bovina y Enfermedad de Creutzfeld-Jakob
El Comité de Redacción de Acta Bioquímica Clínica Latinoamericana ha preparado esta información a partir de dos artículos aparecidos en Mundo científico (julio 2001) y Newton, el espectáculo de la ciencia (marzo 2001) y lo ha compaginado para su difusión a través de FABA-Informa |
|
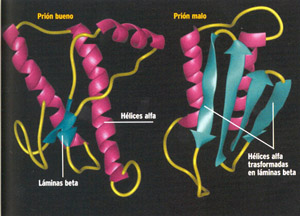
La Encefalopatía Espongiforme Bovina (EEB), es una de las enfermedades que se encuadran dentro del grupo de las llamadas encefalopatías espongiformes transmisibles (EETs). Todas las enfermedades de este grupo poseen una serie de características comunes tales como el largo periodo de incubación que transcurre desde la entrada del agente causal hasta la aparición de las primeras manifestaciones clínicas, el curso progresivo y crónico que ineludiblemente conduce a un desenlace fatal y unas características clínicas y anatomopatológicas definidas y muy similares entre sí en las distintas especies, que tienen en común la afección del sistema nervioso central. En el tejido se detectan la proteína PrPsc, que se relaciona estrechamente con el agente responsable de la enfermedad y las llamadas fibrillas SAF. Las enfermedades del grupo tienen un carácter transmisible. El agente causal es extraordinariamente resistente a tratamientos convencionales y no desencadena reacción inmune. Aunque la naturaleza de los agentes responsables ha sido muy debatida, la hipótesis más aceptada hasta este momento es la que identifica estos agentes como priones. Epidemiología La EEB fue diagnosticada por primera vez en el Reino Unido en 1986, país en el que hasta este momento se han diagnosticado 180.725 casos. La exposición de la población bovina británica habría comenzado en el invierno de 1981-82. Los primeros casos se registraron en 1985, y desde entonces el número de casos confirmados experimentó un progresivo crecimiento que alcanzó su pico máximo en los meses de diciembre de 1992 y enero de 1993. En España el número de casos confirmados es de 33. Los animales afectados tenían edades comprendidas entre los tres años y medio y los catorce años y medio. Los animales corresponden a razas mayoritariamente de producción lechera aunque, en algún caso concreto, eran de aptitud mixta o de producción de carne. Desde el 1 de enero de 2001 se han llevado a cabo un número importante de tests rápidos (entorno a 25.000), de los cuales más de la mitad se ha realizado en Galicia. La proporción de animales positivos en relación con el número de análisis realizados es de 1 positivo por cada 1000 animales analizados. Todos los casos fueron confirmados por el Laboratorio Nacional de Referencia de Encefalopatías Espongiformes Transmisibles de Zaragoza. La enfermedad ha sido también diagnosticada en otros países como Irlanda, Portugal, Suiza y Francia. Otros países donde se ha registrado la enfermedad, pero alcanzando cifras de vacas afectadas notablemente inferiores (entre 1 y 50 casos) son: Alemania, España, Bélgica, Holanda, Dinamarca, Italia, Liechtenstein, Luxemburgo. Fuera de Europa, y debido a la importación de animales que portaban el agente causal de manera inaparente, se han descrito casos en Canadá, Islas Malvinas y el Sultanato de Omán. Hasta el momento no se tiene conocimiento de la existencia de casos en otros países o continentes, además de los referidos. La enfermedad se ha descrito tanto en ganado nativo como en vacunos importados del Reino Unido. Diagnóstico El diagnóstico de la enfermedad se efectúa tomando como base la observación de los síntomas clínicos y su confirmación se realiza mediante el estudio histopatológico de muestras de encéfalo y la observación de las vacuolizaciones neuronales y del neuropilo, características de la enfermedad y la detección de la PrPsc. Cuadro Clínico La EEB se presenta clínicamente en animales adultos de ambos sexos y preferentemente en explotaciones de ganado lechero. El período de incubación es variable pero en general largo, considerándose por término medio en torno a 4-5 años. El curso clínico es progresivo y prolongado, entre 7 y 14 meses. El desenlace es siempre fatal. Los síntomas clínicos de la EEB incluyen cambios en el comportamiento, sensoriales, locomotores y cambios generales. Entre los cambios en el comportamiento se citan el estado de nerviosismo que presenta el animal, desconfianza, hiperestesia, que se manifiesta bajo la forma de respuestas exageradas a estímulos auditivos y táctiles, aprensión, comportamiento agresivo, rechinar de dientes, lameteos frecuentes, movimientos circulares y rápidos de los ojos y reticencia a sortear obstáculos. En cuanto a los cambios sensoriales y locomotores se describen temblores, mioclonias, posturas anormales de la cabeza, movimientos anormales de los pabellones auditivos, hipermetría, incoordinación de movimientos, ataxia, dificultades para mantenerse en la posición habitual, caídas al suelo frecuentes e injustificadas, dificultad para incorporarse y finalmente un estado de gran deterioro neurológico conduce a un estado de postración permanente y a la muerte del animal. El prurito, que es una manifestación común en el Scrapie ovino, no lo es tanto en el bovino. En cuanto a los signos generales, se observa una pérdida rápida de la condición corporal, a pesar de mantener el apetito y una disminución progresiva de la producción láctea. Lesiones Las lesiones se corresponden con la característica de una encefalopatía espongiforme. No son visibles macroscópicamente y no tienen carácter inflamatorio. Se localizan en el sistema nervioso central, sobre todo a nivel de la médula oblongada, puente y mesencéfalo y de forma predominante en el núcleo dorsal del vago, en el tracto solitario, tracto espinal del nervio trigémico, núcleos vestibulares y en la formación reticular. Son lesiones bilaterales y simétricas y consisten en una vacuolización del pericarion neuronal, así como del neuropilo, de los centros referidos. Demostración de la PrPsc Ante un animal que ha presentado síntomas sospechosos de la enfermedad y cuyo perfil lesional concuerda con el patrón característico de la EEB resulta indispensable llevar a cabo la demostración de la proteína PrP patológica (PrPsc) por métodos inmunocitoquímicos en el propio tejido nervioso o mediante otras técnicas como el inmunoblotting o un ELISA. La demostración de la proteína PrPsc mediante la utilización de las dos últimas técnicas citadas, ha supuesto un avance importante en la tecnología diagnóstica de la enfermedad, ya que está permitiendo acortar notablemente el período de tiempo para la identificación de los casos positivos y el procesado y análisis de un importante volumen de animales, lo que resulta de todo punto necesario para el conocimiento de la prevalencia real de la enfermedad en un país. Estos test tras ser sometidos a un proceso de validación por parte de la Comisión Europea han demostrado ser muy sensibles y específicos, y por lo tanto fiables. No obstante, las técnicas de referencia de la enfermedad, de acuerdo al criterio expresado por la OIE y la propia Comisión Europea, siguen siendo la confirmación del perfil lesional y la demostración de la PrPsc con el método de la inmunocitoquímica, que se considera altamente específico y sensible. La enfermedad puede ser confirmada también mediante la inoculación a animales de laboratorio transformados genéticamente (ratones o hámsters), aunque requiere un período de tiempo más prolongado debido a la duración del período de incubación. En la actualidad se están realizando esfuerzos para disponer de pruebas que permitan el diagnóstico de la enfermedad in vivo y en estadios más tempranos, lo que supondría un significativo avance para su control y erradicación. Resulta obligado realizar un diagnóstico diferencial con otras enfermedades que también son responsables de síntomas nerviosos como la hipomagnesemia, listeriosis, cetosis, necrosis cerebrocortical, encefalopatías hepatógenas, rabia, en los países donde existe, entre otras. ¿Qué ocurre en el hombre? Aquí es donde entra en juego la temible enfermedad de Creutzfeldt-Jakob. Bautizada con el nombre de los dos científicos alemanes que la descubrieron en los años veinte, es una dolencia muy rara y con numerosas variantes. La clásica, por ejemplo, afecta tanto a los hombres como a las mujeres de edades comprendidas entres los 60 y los 70 años. Es letal y actúa rápidamente: desde el momento en que aparecen los primeros síntomas hasta la muerte transcurren, de media, seis meses. Durante ese periodo, la persona afectada empieza a sufrir pérdidas de memoria y desorientación espacial y temporal; luego, falta de coordinación de los movimientos, alucinaciones e incapacidad manifiesta para reconocer los objetos, aun cuando no haya lesiones en los ojos. En la fase final, la persona deja de hablar y no se vuelve a mover hasta su muerte. Otra variedad de la enfermedad Creutzfeldt-Jakob, prácticamente desaparecida, es conocida como Kuru. Se originó hace años en Papua Nueva Guinea, en una comunidad que practicaba el canibalismo. Además de los síntomas clásicos, estos individuos padecían un característico temblor (kuru, en su lengua) que les impedía la coordinación de los movimientos y provocaba violentos cambios de humor. De esta enfermedad existen, también, formas hereditarias, como el insomnio fatal o la que ahora está relacionada con las vacas locas. Tanto en los casos de formas hereditarias de la enfermedad de Creutzfeldt-Jakob, como en los casos de vaca loca, la proteína priónica normal, que existe en todos los mamíferos, muta y se vuelve devastadora para el sistema nervioso. Una mutación misteriosa Por qué ocurre esto sigue siendo un misterio. La teoría más acreditada es la que propuso, en 1997, el Premio Nobel estadounidense Stanley Prusiner. Según este investigador de la Universidad de San Francisco, en California, la proteína priónica se vuelve loca espontáneamente, muta y se transforma en una especie de molde en el que las proteínas buenas, producidas normalmente por las células, se amoldan para convertirse a su vez en malas. Algo similar al efecto dominó: se golpea una ficha y todas las demás caen irremediablemente. Más incógnitas. Todavía no queda claro si el daño generado en el tejido cerebral se debe a la acumulación de proteínas malas o a un efecto tóxico de las mismas. La teoría plantea dudas. Podría existir un virus que se une a la proteína priónica buena , y gracias a ella entra en la célula y la transforma en mala. Las implicaciones futuras relativas a estas dos teorías son enormes. Si la hipótesis del prión fuera correcta, ello querría decir que el único sistema para hacer un diagnóstico está condicionado por la individuación de la proteína mala. Si por el contrario, se tratara de un virus, sería posible identificarlo, puesto que la investigación para descubrir nuevos virus está muy desarrollada en todo el mundo. En busca del nexo Pero ¿será cierta la conexión entre el síndrome de Creutzfeldt-Jakob y el consumo de carne de vaca loca? Gracias a los estudios hechos desde 1993 en Europa, Canadá y Australia, se han podido establecer las características de la enfermedad. La mayor parte de los pacientes detectados hasta hoy eran, curiosamente, jóvenes menores de 30 años, lo que en cierto modo echa por tierra la idea de que la enfermedad Creutzfeldt-Jakob afecta, sobre todo, a personas mayores de 50 años. En todos los pacientes examinados, la mayoría en Gran Bretaña, la dolencia era más larga, duraba más de seis meses. Esto ha permitido avanzar la hipótesis de un nexo entre la nueva enfermedad y el mal de las vacas locas. Y un dato más: se le ha inyectado material cerebral humano a ratones y se ha controlado no sólo el tiempo que tardaban en enfermar sino las partes de su cerebro que quedaban afectadas. Pues bien, los ratones presentaban el mismo período de incubación y el mismo tipo de lesiones cerebrales que aquellos que habían sido infectados con el material extraído de las vacas locas. El origen del mal Aunque sus efectos son de sobra conocidos, no todos los expertos coinciden a la hora de establecer los orígenes de la enfermedad. Hasta hace tan solo unos meses se creía que el agente infeccioso de la EEB, un prión, procedía de otra proteína que causaba en los ovinos un tipo de Encefalopatía Espongiforme conocida desde el siglo XVII. El contagio pudo haberse producido en Gran Bretaña a través de unos esqueletos de ovejas infectadas, introducidas accidentalmente en los procesos de elaboración de las harinas de carne para la alimentación de ganado. Todo esto habría ocurrido en un momento en el que la industria, con el ánimo de ahorrar energía, había bajado las temperaturas de elaboración recomendadas (133º C) para eliminar el agente infectante de los piensos. Por otra parte, la comisión científica que asesora al Parlamento inglés ha revalorizado la teoría de la mutación genética del prión, la proteína que se encuentra, en forma inocua, en todos los mamíferos, incluido el hombre. Un solo animal con el prión mutado introducido en la producción de harinas de carne, habría provocado la epidemia. Pero una cosa es el origen del agente infectante (para nada claro), y otra, su difusión, debida seguramente a los piensos cárnicos utilizados para alimentar a las reses. Qué nos espera ¿Podrían hacerse previsiones acerca de cómo la enfermedad se manifestará en el futuro? Ninguno de los científicos consultados se ha arriesgado a dar un pronóstico. La solución dependerá mucho de los controles sanitarios que se están haciendo, independientemente de los resultados que se obtengan de las investigaciones. Nadie, en definitiva, está en condiciones de hacer previsiones numéricas acertadas. Entre otras cosas, porque no se conocen los tiempos de incubación de la enfermedad. Cuando se habla de un período de entre 5 y 30 años se toma como modelo el Kuru que, por otra parte, es un ejemplo de transmisión de hombre a hombre. En nuestro caso, por el contrario, está en juego la llamada barrera de especie, es decir, aquella que se puede instaurar entre el animal y el hombre. Pero no se sabe nada más. Por otra parte, no existe ningún test que permita identificar las infecciones en humanos hasta que no se hayan manifestado los primeros síntomas. La proteína escurridiza El hallazgo de los priones, las proteínas que causan un tipo de enfermedades conocidas genéricamente como Encefalopatías Espongiformes Transmisibles (EET), es un ejemplo de cómo las certezas científicas pueden derrumbarse frente a la realidad. Hasta su descubrimiento, realizado en 1984 por el norteamericano Stanley Prusiner, cada investigador habría jurado que para provocar una enfermedad infecciosa son necesarios organismos dotados de ADN o de ARN, es decir, de elementos capaces de autorreplicarse en el interior del huésped: por ejemplo, virus, parásitos u hongos. Pero los priones son diferentes: no tienen material genético, sólo proteínas. Ahora los investigadores han tenido que reconocer que cada una de estas proteínas dispone de la capacidad suficiente para burlar las defensas de sus víctimas e infectar su cerebro. Son por así decirlo, los priones malos, formas aberrantes que atacan rápidamente al cerebro paralizando la transmisión entre las células nerviosas. ¿Podría estar el mal en la leche que tomamos? Parece una perspectiva aterradora. Sin embargo, aseguran los expertos, la probabilidad es mínima. Al día de hoy "no se han encontrado pruebas de que la Encefalopatía Espongiforme Bovina (EEB) se transmita a través de la leche de las vacas infectadas por priones", asegura Víctor Briones, Director del Departamento de Sanidad Animal de la Facultad de Veterinaria de la Universidad Complutense. Sin embargo, puntualiza, ?el riesgo cero no existe?. Tampoco el profesor Peter Smith, máxima autoridad británica sobre la enfermedad de las vacas locas, se atreve a dar una respuesta definitiva. "Nadie, ni el más sabio, puede descartar rotundamente este peligro". Y añade, "lo único que se puede decir al respecto es que en la actualidad hay varias investigaciones abiertas para tratar de averiguar si hay la más mínima posibilidad de que la leche sea un material peligroso. Y por ahora, no se han obtenido datos sospechosos". La propia Organización Mundial de la Salud (OMS) está estudiando la necesidad de actualizar sus recomendaciones sobre los riesgos de productos como la leche, las gelatinas y las grasas procedentes de animales con EEB. Puestos en el peor de los casos, sería difícil, por no decir imposible, dar con un sustituto ideal de la leche natural. Entre otras cosas porque, según los expertos en nutrición, ningún otro producto (natural o sintético) reúne tanta cantidad y variedad de vitaminas, minerales, proteínas y ácidos grasos necesarios. De hecho, la leche de vaca, es el alimento más consumido en el mundo. Si faltase, habría que echar mano de los preparados de laboratorio como la leche de soja o de almendra. ¿Cuánta carne hay que comer para contraer la enfermedad? Bastaría un solo gramo de tejido cerebral para infectar a un animal. Una persona necesitaría ... Seguramente nos hemos preguntado más de una vez que cantidad de carne tendríamos que comer para adquirir la enfermedad de Creutzfeldt-Jakob, es decir, la variante humana del mal de las vacas locas. Y es que durante todos estos años anteriores prácticamente nadie ha dejado de ingerir carne. Pues bien, tampoco en este caso los científicos tienen una respuesta clara ni mucho menos tranquilizadora. Lo que sí se sabe es que un solo gramo de tejido cerebral, el más infeccioso, es suficiente para contagiar a una vaca o a una oveja y matarlas. "Ocurre lo mismo en los seres humanos" No se tiene idea. Los expertos confían en que las variaciones que se producen cuando una enfermedad salta de una especie a otra haga necesario dosis mucho más altas de tejidos enfermos para contraer el mal. Lo único que se sabe es que las personas afectadas en Gran Bretaña eran consumidoras habituales de carne de vacuno. Además, "tampoco sabemos cuál es el período de incubación de la enfermedad", explicaba hace unos días el epidemiólogo británico Peter Smith. Se habla de entre 10 y 15 años. Él sospecha no sólo de la carne de bovino sino de otros productos de mala calidad como salchichas y hamburguesas en las que "probablemente pueda encontrarse el agente infeccioso". Las estimaciones que barajan los expertos, entre ellos el profesor Smith, no son precisamente tranquilizadoras en cuanto al número de afectados previsto de aquí a los próximos años. Para Smith, "es posible que estemos en los inicios de una epidemia que se prolongue durante mucho tiempo" y que afectará sólo en Gran Bretaña, a 100.000 personas, según algunos cálculos. "Y en España" Oficialmente no se ha producido ningún caso en humanos, a pesar de que el número de animales con Encefalopatía Espongiforme Bovina (también los toros de lidia están bajo sospecha) sigue aumentando en todo el territorio. La erradicación de la enfermedad, según el doctor Víctor Briones, sería viable en el plazo de un año. |